News



On January 29,2026,the research group led by Associate Professor Haobin Ye published a research article entitled “Dual Targeting of SLC25A51 and Succinate Dehydrogenase Selectively Depletes Mitochondrial NAD+ to Eradicate KRAS-Driven AML” in Cell Metabolism.

Acute myeloid leukemia (AML) arises from diverse mutations, yet its most aggressive drivers remain elusive. Here, we show that Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations drive hyperproliferative and therapy-/glucose stress-resistant AML, whereas existing inhibitors lack sufficient cytotoxicity. Dual physiological/glucose-deprived screening identified compound 615 selectively eliminating KRAS-mutant cells through concurrently inhibiting succinate dehydrogenase (SDH) and the cytosol-to-mitochondrial NAD+ transporter SLC25A51. Mechanistically, KRAS-mutant cells exhibit reduced 2-oxoglutarate dehydrogenase complex-mediated SLC25A51 K264 succinylation, a mitochondrial NAD+-dependent modification promoting protein stability. This creates a synthetic lethal vulnerability: low-dose 615 triggers a cascade failure by acutely inhibiting SLC25A51, followed by its destabilization, causing complete transporter suppression. Together with concurrent SDH inhibition, this drives catastrophic mitochondrial NAD+ depletion. Conversely, KRAS-wild-type cells preserve NAD+ influx via sufficient baseline succinyl-SLC25A51, which stabilizes SLC25A51 and enables sufficient succinate accumulation to drive hypoxia inducible factor 1 subunit alpha (HIF1α)-mediated compensatory NAD+ production during treatment. Our work reveals a KRAS-specific metabolic vulnerability and proposes a dual-inhibition therapy for KRAS-driven AML.
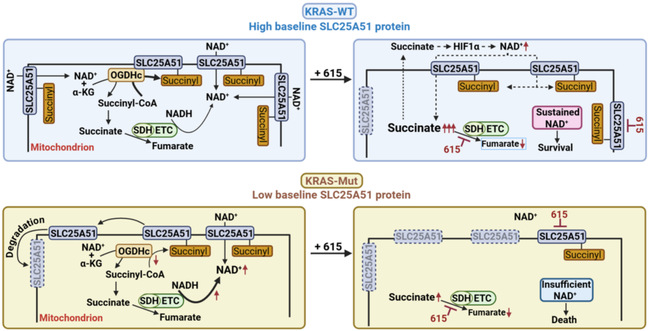
Figure: Dual inhibition of SLC25A51 and SDH depletes mitochondrial NAD+ and selectively eliminates KRAS-mutated acute myeloid leukemia cells
Link: https://doi.org/10.1016/j.cmet.2026.01.001

复旦大学 代谢与整合生物学研究院 版权所有
Copyright@2018fudan.edu.cn All rights reserved.
上海市杨浦区淞沪路2005号。邮编:200438。电话:021-31242078,021-31242079
 Fudan Email
Fudan Email Fudan Ehall
Fudan Ehall