
新闻中心


2024年2月12日,复旦大学代谢与整合生物学研究院青年研究员任若冰课题组联合香港中文大学(深圳)助理教授竺立哲团队、2013年诺贝尔化学奖得主南加州大学Arieh Warshel教授在国际学术期刊The Proceedings of the National Academy of Sciences在线发表了题为Fine-tuning activation specificity of G-protein-coupled receptors via automated path searching的研究成果。
G蛋白偶联受体(GPCR)是大约35%的美国食品药品监督管理局批准的药物的靶点。然而,设计选择性GPCR激动剂仍具有挑战性,因为需要深入了解各种配体对多种GPCR亚型的激活机制。此外,GPCR亚家族成员之间的序列高度相似性对选择性激动剂的设计提出了严峻的挑战。例如,1-磷酸鞘氨醇受体(S1PR)家族的五个成员在其激动剂结合口袋具有约70%的序列相似性(图1)。目前,大多数S1PR激动剂,如芬戈莫德磷酸盐(FTY720P)、CBP-307和西波尼莫德(BAF-312),会同时激活多种亚型S1PRs(图1C)。当内源性S1P信号在疾病治疗中被不可区分地劫持时,这种混杂性破坏了这些配体在扩展适应症和减少副作用方面的功效,因此迫切需要设计S1PR1的高选择性激动剂。

图1 A类GPCR激动剂设计中的特异性问题
精细的GPCR激动剂设计需要对激活机制有深入的了解,这对现有技术来说是一项艰巨的任务。结构生物学技术通常为非活性受体和活性受体-配体复合物提供宝贵的原子结构,但较难捕获关键的、短暂的激活过渡状态。分子动力学模拟可以提供精细的活化细节,但效率较低。所以,迫切需要一种高效率计算方法指导高选择性配体分子药物设计。在这篇文章中,研究者们描述了一种自动化无输入路径搜索方法(图2),该方法可以找到任意配体激活鞘氨醇-1-磷酸受体(S1PR)的过程在高维空间中定义的最小自由能路径(MFEP),从而预测配体的效力,并揭示受体的激活过程。结果显示,在4个不同配体和3个S1PR亚型的两两组合中,沿MFEP的自由能分布与G蛋白解离的生物发光共振能量转移(BRET)测定结果达到了显著的一致。特别的是,计算得到的过渡态结构揭示了S1PR3残基F263/I284是决定现有激动剂CBP307和BAF312特异性激活S1PR1/5的关键残基。BRET检测证明,在S1PR1和S1PR3之间进行这些残基的互换突变会逆转两种受体对现有激动剂的响应。这些结果启发研究者们,具有强极性头部和大型疏水尾部的激动剂可能会具有对S1PR1更高的选择性。因此,该研究针对S1PR1进行了新型激动剂设计。仅通过两次迭代,研究者的计算工具即预测出了一种新型S1PR1特异性激动剂。BRET测定证实,新型分子的两种手性形式均能在纳摩尔浓度下激活S1PR1,该浓度比激活S1PR3/5所需浓度低2个数量级。
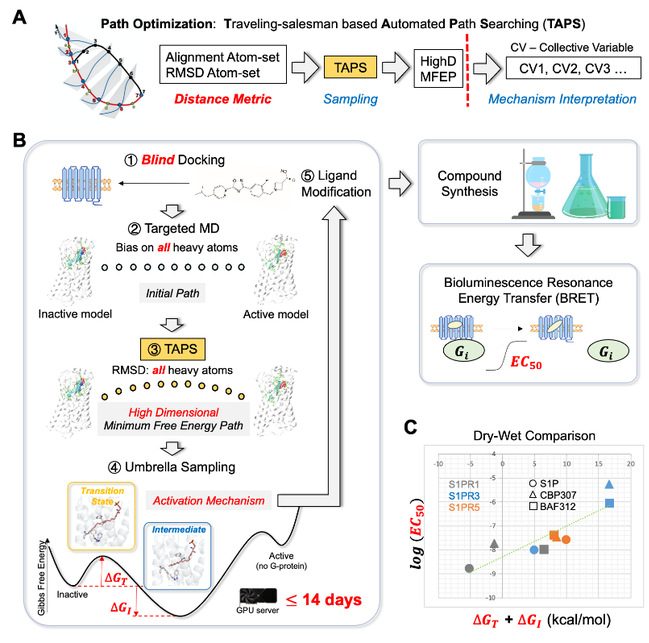
图2 用于GPCR激动剂合理设计的激活路径的自动搜索算法
综上所述,基于该搜索路径,使开发具有更高选择性和高效性的靶向S1PR的药物小分子成为可能。结果也表明,研究者开发的计算方法在G蛋白偶联受体激动剂设计方面具有很好的应用前景。
本研究中,竺立哲课题组博士生题如涓,任若冰课题组博士生庞滨为本文共同第一作者。复旦大学代谢与整合生物学研究院博士后玉蕾叶、香港中文大学(深圳)博士生甘冰、马文卓也做出了贡献。
论文链接:
www.pnas.org/doi/10.1073/pnas.2317893121

复旦大学 代谢与整合生物学研究院 版权所有
Copyright@2018fudan.edu.cn All rights reserved.
上海市杨浦区淞沪路2005号。邮编:200438。电话:31242078,31242079
 复旦大学网上办事服务大厅
复旦大学网上办事服务大厅 复旦大学实验室安全教育与管理平台
复旦大学实验室安全教育与管理平台