
新闻中心


TBCK综合征是一类由编码TBC1结构域蛋白激酶(TBCK)的基因突变引起的罕见的遗传性神经发育障碍疾病,患者通常表现为发育迟缓、智力障碍、肌张力低下,部分伴随癫痫和多系统损害,但其具体的发病机制长期不明。
2025年11月8日,复旦大学附属妇产科医院/代谢与整合生物学研究院王红艳及其合作团队在Science Bulletin期刊发表了题为“TBCK syndrome pathogenesis: the TBCK-PPP1R21-C12orf4 complex regulating RAB5-dependent endo-lysosomal homeostasis”的研究论文,对TBCK基因突变导致TBCK综合征(TBCK syndrome)的致病机理进行了深入探索。本研究综合大规模基因共依赖性分析、蛋白互作组学、基因编辑细胞与基因敲除小鼠模型等技术手段,鉴定到TBCK与PPP1R21、C12orf4能够形成稳定的三元蛋白复合体。该复合体与小GTP酶RAB5直接作用并行使潜在的Rab GAP功能,进而抑制RAB5活性,并且共同维持内吞-溶酶体系统稳态。而PPP1R21和C12orf4基因失活突变也曾被报道能够同样导致类似的神经发育障碍,这种共病现象的内在联系尚待阐明。

研究团队揭示,TBCK缺失或Rab GAP-TBCK结构域的活性位点错义突变均会导致TBCK-PPP1R21-C12orf4三元复合体装配受损,进而引发RAB5持续性过度激活。机制上,研究团队首次证明,TBCK基因内含直接抑制RAB5活性的GAP结构域,而PPP1R21和C12orf4是发挥GAP活性的结构性伴侣蛋白。RAB5作为早期内体成熟和囊泡运输的核心调控因子,当其活性异常增强时,会激活一系列下游效应蛋白,造成早期内体显著扩大和向晚期内体转换过程受阻,进而导致内吞降解、溶酶体和自噬等异常,最终破坏细胞的内吞-溶酶体稳态。PPP1R21或C12orf4缺失亦导致类似的异常细胞表型。这些病理性改变不仅在体外细胞实验中得到证实,在Tbck、Ppp1r21和C12orf4基因敲除小鼠中,也观察到高度一致的现象。值得注意的是,Tbck或Ppp1r21缺失小鼠表现为围产期纯合致死,而C12orf4缺失小鼠则能存活并表现出显著的认知障碍与运动功能缺陷,其脑部MRI和行为学测试结果与临床患者高度相似,从而为临床症状提供了有力的动物学佐证。
这一系列发现描绘了TBCK综合征的全新发病图景:TBCK-PPP1R21-C12orf4复合物行使RAB5的GTP酶活负调控因子的角色,在神经发育过程中维持内吞、溶酶体与自噬的平衡;当该复合物功能受损时,细胞内体-溶酶体系统稳态被打破,进而导致神经元结构与功能障碍,并引发一系列临床表型。该研究不仅阐明了TBCK-PPP1R21-C12orf4复合体亚基失活突变致病的分子机制,同时提示,靶向降低RAB5活性是治疗这类罕见病的潜在干预策略,为精准医学和药物研发提供了新方向。
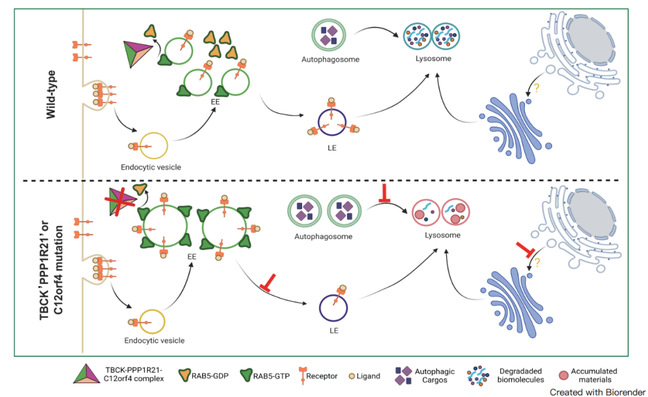
图1:TBCK-PPP1R21-C12orf4复合物破坏导致RAB5过度激活与内体-溶酶体稳态异常
复旦大学附属妇产科医院/代谢与整合生物学研究院王红艳教授和生命科学学院王陈继副研究员为该论文的共同通讯作者,复旦大学代谢与整合生物学研究院博士后王亚兰和生命科学学院博士生徐夏韵为共同第一作者。该工作得到了复旦大学生命科学学院麻锦彪教授在蛋白纯化过程中的鼎力帮助,受到国家自然科学基金(32488101、32370726、92357301等)、国家重点研发计划(2022YFA1104200)、上海市自然科学基金(22ZR1406600)、深圳市医学研究基金(B2402004)、深圳市“三名工程”项目(SZSM202311005)以及上海市科技计划等项目的大力支持。
原文链接:
https://www.sciencedirect.com/science/article/pii/S2095927325010576

复旦大学 代谢与整合生物学研究院 版权所有
Copyright@2018fudan.edu.cn All rights reserved.
上海市杨浦区淞沪路2005号。邮编:200438。电话:31242078,31242079
 复旦大学网上办事服务大厅
复旦大学网上办事服务大厅 复旦大学实验室安全教育与管理平台
复旦大学实验室安全教育与管理平台