
新闻中心


2020年2月,学术期刊Cell Research发表了由复旦大学张锋课题组、华克勤课题组、中科院生化与细胞所李劲松课题组共同牵头的女性苗勒管发育不良(Müllerian anomalies, MA)的最新研究成果:提出并证明两个或多个基因突变的协同叠加效应是导致MA的重要原因,该研究将引领和激励临床和基础相关人员从人类复杂疾病中搜寻更多的致病性基因突变组合。
对于复杂人类疾病来说,单基因突变通常很难解释疾病的发生。女性苗勒管发育不良是女性生殖系统(子宫、宫颈、阴道等)发育畸形的统称,也常合并泌尿系统发育异常,是一个复杂的先天性疾病。在临床上,MA患者表现为严重程度不一的生殖系统形态结构异常,如无子宫无阴道、单角子宫、残角子宫、宫颈阴道闭锁等等。多年来,各国研究人员尝试寻找MA的遗传致病机制,然而进展缓慢。多数的MA病例无法用单基因模式来解释,这提示该病的遗传复杂性以及探究双基因(digenic)或者寡基因(oligogenic)模式的急迫性。
研究团队收集了125例苗勒管发育不良的患者血液样本,首先对其进行表型分型;随后利用qPCR、全基因组杂交芯片对样本进行了分析,发现9例携带有罕见的基因组拷贝数变异(copy number variants, CNVs);然后利用家系遗传分析发现部分患者的母亲携带相应的CNV。这些结果提示单一的遗传变异不足以导致苗勒管发育不良(图1)。因此,研究人员进一步利用全外显子测序技术对上述9例CNV携带者进行了全外显子测序,结果发现其中1例GEN1/2p24.2缺失患者还携带有WNT9B基因突变、1例TBX6/16p11.2缺失患者还携带有GATA3基因突变。由此推测“GEN1+WNT9B”、“TBX6+GATA3”这2个突变组合可能是导致人类MA的致病原因。

图1 双/寡基因突变导致MA的模式图
研究团队进一步利用半克隆技术结合CRISPR-Cas9基因编辑技术构建了多基因突变小鼠模型,使得上述研究结果在小鼠动物模型上得到了科学、有利的论证。精子来源的孤雄单倍体胚胎干细胞(AG-haESCs)可以在体外长期培养,通过流式分选可以稳定地维持单倍体细胞的存在。利用CRISPR-Cas9技术对AG-haESCs进行多基因编辑,随后显微注射至卵母细胞中并胚胎移植至假孕小鼠体内,可以一步获得多基因突变的小鼠模型(半克隆技术)。该技术方法效率高、周期短。研究团队利用上述半克隆技术、CRISPR-Cas9基因编辑技术快速构建了“GEN1+WNT9B”、“TBX6+GATA3”双突变小鼠模型(图2)。通过后续的一系列分析发现,“GEN1+WNT9B”突变组合可以导致小鼠苗勒管发育不良。在MA家系和小鼠模型中,GEN1或WNT9B单基因突变都不足以致病,但是两个基因突变的叠加则具有致病性。综上所述,MA患者和小鼠模型的分析结果不仅证明了“GEN1+WNT9B”双基因突变组合是导致MA的致病原因,还展示了半克隆技术结合CRISPR-Cas9基因编辑技术在复杂人类疾病研究中的应用。
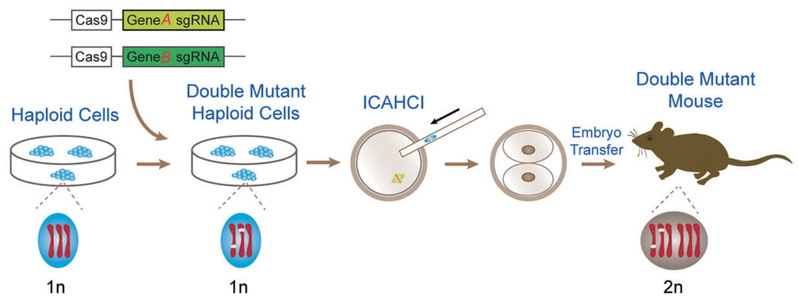
图2 半克隆技术结合CRISPR-Cas9快速制备双/寡基因突变小鼠的流程图
论文链接:

复旦大学 代谢与整合生物学研究院 版权所有
Copyright@2018fudan.edu.cn All rights reserved.
上海市杨浦区淞沪路2005号。邮编:200438。电话:31242078,31242079
 复旦大学网上办事服务大厅
复旦大学网上办事服务大厅 复旦大学实验室安全教育与管理平台
复旦大学实验室安全教育与管理平台