
新闻中心


2026年4月10日,复旦大学代谢与整合生物学研究院任若冰课题组联合上海交通大学附属第六人民医院玉蕾叶助理研究员在国际知名期刊PLoS Biology在线发表了题为“Structural insights into subtype-specific agonist recognition by Sphingosine-1-phospate receptors”的研究论文。该工作整合结构生物学及分子动力学模拟与系统的药理学分析,系统揭示了四种鞘氨醇-1-磷酸受体激动剂——CYM5442、HYX1011、Ponesimod和SAR247799高选择性激活S1PR1的分子机制,为理性设计更安全、更高效的新一代靶向S1PR1小分子药物提供了关键的结构基础与理论指导。

鞘氨醇1-磷酸受体(Sphingosine-1-phosphate receptors,S1PRs)是一类重要的A类G蛋白偶联受体(GPCR),包括五种受体亚型(S1PR1-S1PR5)。S1PRs激活介导的下游信号通路淋巴细胞迁移、血管内皮屏障稳态、中枢神经系统功能调控等生理过程中发挥核心作用,并广泛参与心血管疾病、自身免疫病、炎症反应、神经退行性疾病、肿瘤进展、听力损伤及纤维化等多种病理过程。其中,S1PR1已被确立为多发性硬化症(MS)和炎症性肠病(IBD)等自身免疫性疾病的优选治疗靶点。目前临床应用的代表性药物,如第一代S1PR调节剂芬戈莫德(Fingolimod/FTY720)及其衍生物西尼莫德(Siponimod),虽具显著疗效,但普遍存在多亚型激活特性,从而引发非预期的下游信号激活,导致心动过缓等相关不良反应,严重制约其长期用药的安全性与耐受性。
开发高度选择性靶向S1PR1亚型的小分子激动剂/调节剂,是突破当前临床药物分子局限的重要路径。然而,S1PR1-S1PR5五种亚型在氨基酸序列、三维结构及配体结合口袋等方面高度保守,尤其在正构结合位点区域相似性极高,使得实现受体单一亚型精准识别面临巨大挑战。因此,深入解析激动剂如何在结构层面“区分”S1PR1与其他亚型,不仅具有重要基础科学意义,更是推动靶向S1PR1精准药物研发的核心前提。
针对这一难题,研究团队选取四种已报道的具有不同程度S1PR1选择性的代表性激动剂,成功解析了其分别与S1PR1-Gi1蛋白复合物的高分辨率冷冻电镜结构。这些结构在原子水平上清晰呈现了不同激动剂与S1PR1的结合模式、关键相互作用位点以及受体激活过程中的构象变化特征。
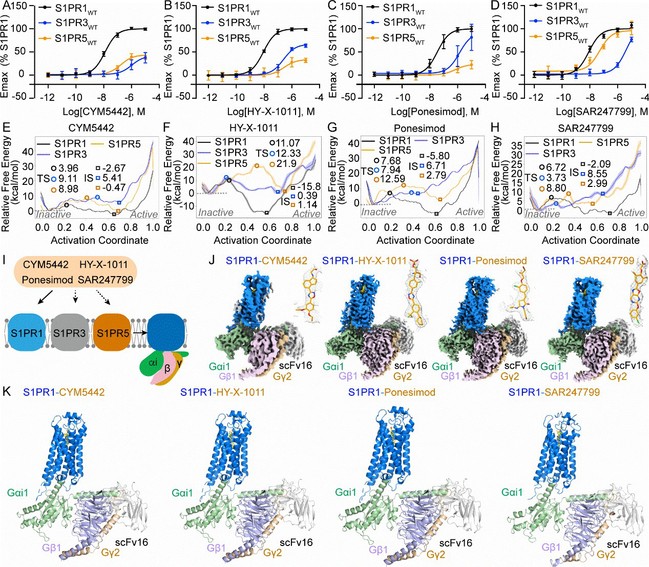
研究发现:
1.非保守氨基酸协同决定选择性:S1PR1配体结合口袋及Gi蛋白偶联界面中多个非保守氨基酸共同提高激动剂对S1PR1的特异性识别和激活能力;
2.空间位阻调控亚型偏好:激动剂分子靠近跨膜螺旋TM5–TM7区域的结合取向,配合其尾部引入的较大分支取代基,与S1PR3中对应位置的非保守大侧链氨基酸发生显著空间位阻冲突,从而阻碍其有效结合;此外,激动剂尾部较大的支链结构会阻碍其深入插入S1PR5的配体结合口袋底部,从而限制其对这种亚型的高效激活。
3.保守氨基酸的影响:激动剂与结合口袋顶部高度保守的极性氨基酸所形成的极性相互作用,也在调控亚型选择性中发挥作用。
4.口袋拓扑差异限制结合兼容性:S1PR2与S1PR4的配体结合口袋上部因存在侧链较大的非保守氨基酸,导致口袋入口显著收窄,使具有较宽分子轮廓的S1PR1选择性激动剂难以进入并稳定结合。
综上,该研究从结构生物学角度系统阐明了S1PR1选择性激动剂实现亚型识别的多层次分子逻辑,构建了基于结构的S1PR1高选择性激动剂的设计框架。研究成果不仅深化了对S1PRs家族配体识别机制的理解,也为后续开展基于结构的药物优化和理性化药物设计、人工智能辅助分子生成及靶向S1PR1亚型选择性调控策略提供了坚实支撑。未来,有望加速开发新一代低脱靶风险、高治疗指数的S1PR1靶向候选药物,助力相关疾病治疗药物分子的精准化升级。
复旦大学代谢与整合生物学研究院博士后玉蕾叶(现任上海市第六人民医院助理研究员)、香港中文大学(深圳)焦海展博士、复旦大学代谢与整合生物学研究院庞滨博士和香港中文大学(深圳)题如涓博士为本文共同第一作者,复旦大学代谢与整合生物学研究院任若冰研究员和玉蕾叶博士为本文的共同通讯作者。复旦大学代谢与整合生物学研究院博士后甘冰博士、硕士研究生秦朝阳、香港中文大学(深圳)助理教授竺立哲和胡红丽以及第二军医大学药学院王金鑫教授参与部分研究或者提供了相关支持。本研究得到中国博士后科学基金面上项目的支持。
原文链接:
https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.3003381

复旦大学 代谢与整合生物学研究院 版权所有
Copyright@2018fudan.edu.cn All rights reserved.
上海市杨浦区淞沪路2005号。邮编:200438。电话:31242078,31242079
 复旦大学网上办事服务大厅
复旦大学网上办事服务大厅 复旦大学实验室安全教育与管理平台
复旦大学实验室安全教育与管理平台